材料模擬與計算設計是繼實驗研究方法、理論研究方法之後的第三個重要科學研究方法,對未來科學技術的發展將扮演越來越重要的角色。材料的理論模擬與計算設計是結合現代的材料科學與技術、物理學基本原理,以及計算機技術而產生的新興學科,已經成為當前材料科學研究中最為活躍的熱點之一。
材料電子結構的計算方法基本分為兩類:半經驗的計算方法和第一原理計算( First Principles Calculation )方法。半經驗的計算方法在歸納總結實驗和數據的基礎上,擬合出相似規律的計算參數,並應用到其他的體系上;而第一原理計算方法是指在計算時,除了告訴程序所使用的原子和它們的位置外,沒有其它任何實驗、經驗或者半經驗的參量。它是經由解薛丁格方程式,得到相對應材料的各種宏觀性質。理論上任何材料的結構和性能可由薛丁格方程式求之,因而第一原理計算在材料計算與模擬領域便顯得十分重要。密度泛函理論( Density Functional Theory; DFT )是以電荷密度取代電子波函數作為研究的基本量,通過 Kohn-Sham方程式將多體問題簡化成沒有交互作用的電子在有效場中運動。
建構以第一原理計算的材料電子結構與巨觀性質相關數位數據資料庫為工業材料研發的基礎工作,讓工業材料創新以十倍速增加。因此,2011年美國總統歐巴馬提出「材料基因組計畫」( Materials Genome Initiative; MGI ),其中提出了材料創新需結合三個平台:①計算工具平台;②數位數據資料庫;③實驗工具平台(圖一)。
第一原理計算用來設計新型 OLED分子
台灣 OLED材料開發分散於工研院、學界及業界,但投入的資源都不多,整體來說,落後國外 3~5年。為了建立自主設計與合成能力,產生各色 OLED材料,研究團隊使用 MGI手法,以量子力學理論模擬大量計算 OLED分子片段之 HOMO、LUMO與能隙資料,並建立資料庫之流程如下︰首先以分子力學與半經驗法求出結構最佳化,作為量子力學計算之初始結構,經由量子力學結構最佳化後,再模擬IR頻譜判斷是否為穩定結構。
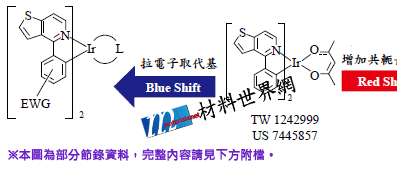
以IR頻譜作為結構穩定之判斷後,進行 HOMO、LUMO與能隙能量之計算。目前建立之資料庫格式如圖五所示,模擬資料庫大約有二百多筆,由模擬的資料庫結果分析得知:增加配位基的苯環共軛長度,可使HOMO上升、LUMO下降,Band Gap減小,導致光色紅移;結構中導入拉電子基團,可使 HOMO下降、LUMO上升,Band Gap增大,導致光色藍移。目前實驗組已將此結果導入工研院材化所開發之 PO-01黃光材料中(圖六),以此結構出發,開發其它光色材料。
圖六、工研院材化所開發之黃光材料PO-01結構
催化劑基因組
一個催化反應並不是單一個因素就能決定結果,在一般的催化反應中,催化劑本身的穩定性、催化劑對於反應的速率、產物的選擇性等數據,都是化學家好奇的,最終目的希望藉由這些數據,根據自訂的篩選順序,找出有潛力的催化劑。
當然有效的催化劑通常不是單一種元素的純金屬,有可能二元或三元合金,以二元合金為例,針對以下四個篩選因子作說明:②反應活性:首先確定反應的所有單元步驟(Elemetary Steps)後,則可根據穩定狀態近似法( Steady-state Approximation ),求出平衡時總反應的速率,就可代表催化劑對於這個反應的活性,平衡時總反應的速率越大,催化劑對於此反應活性越大。圖十一為經過模擬所得的催化劑活性火山圖。④催化劑價格:對於需要大量生產的催化工業,催化劑價格也是考量的重要因素。
圖十一、以理論模擬得到的催化劑活性火山圖
以下為催化劑基因組的例子:由甲醇脫氫催化合成無水甲醛,將不同組合的二元合金材料,透過上述的各項篩選條件,這些二元合金材料首先透過因子穩定性和持久性的篩選,留下來的材料接著進行第二層產物選擇性的篩選,第三層則是---以上為部分節錄資料,完整內容請見下方附檔。
作者:黃瓊慧、李涵榮、邱顯浩 / 工研院材化所
★本文節錄自「工業材料雜誌」361期,更多資料請見下方附檔。